Forschungsbericht 2016 - Max-Planck-Institut für Chemische Physik fester Stoffe
Kovalenz und Ionizität in Verbindungen mit MgAgAs-Struktur: Von Konzepten zur Strukturvorhersage
Das Jahr 2016 war im Bereich der Chemie stark durch den 100. Geburtstag des Lewis’schen Modells der chemischen Bindung geprägt [1]. Interessanterweise ist diese Modellvorstellung zur Elektronenpaarbindung bereits vor Einführung der Quantenmechanik entstanden, rein auf der Basis eines breiten empirischen Wissens um chemische Sachverhalte wie Reaktivität, Zusammensetzungen und des periodischen Systems der Elemente. Aus der anschließenden Pionierzeit der Quantenmechanik gingen zwei Berechnungsverfahren der elektronischen Struktur hervor, die Molekülorbitalmethode (delokalisierter Ansatz) und die Valenz-Bindungs-Methode (lokalisierter Ansatz), die sich zwar in ihrem Grundansatz diametral unterscheiden, jedoch in ihren physikalischen Aussagen zu quantenmechanischen Observablen völlig gleichwertig sind. Die Ursache hierfür ist, dass die gleichen reduzierten Dichtematrizen im Orts- und Impuls-Raum erhalten werden können, die das Verhalten der Elektronen vollständig beschreiben.
Die in der chemischen Begriffswelt so wichtige Identifizierung von Atomen in einem chemischen System wie einem Molekül oder Festkörper gelingt gemäß der QTAIM-Methode (Quantentheorie von Atomen in Molekülen) auf der Basis der einfachsten Dichtematrix, der Einteilchen-Elektronendichte. Atome sind hiernach als Dichtebassin-Regionen im Ortsraum definiert. Sie sind durch Flächen begrenzt, die dem Charakter von geographischen Wasserscheiden entsprechen, d. h. der Fluss der Elektronendichte durch diese Fläche ist Null, und jedes Atom besitzt sein eigenes Elektronendichtebassin, das den Atomkern umschließt. So definierte Atome stellen bewiesenermaßen quantenmechanische Subsysteme dar, was sie von anderen Varianten klar heraushebt. Die Aufteilung des Raumes in QTAIM-Atome ist vollständig, sodass jede Region einem Atom eindeutig zugeordnet ist. QTAIM-Atome besitzen eine Substrukturierung in schalenartige Regionen, die von einigen Realraum-Indikatoren dargestellt wird. Die Chemiker am Max-Planck-Institut für Chemische Physik fester Stoffe (MPI-CPfS) haben sich von Beginn an mit einem derartigen, sehr erfolgreichen Realraum-Indikator, der Elektronen-Lokalisierungsfunktion (ELF) befasst, und diesen durch Einführung des Elektronen-Lokalisierbarkeitsindikators (ELI) für zeitabhängige, korrelierte und relativistische Szenarien verallgemeinert [2]. Auch die Aufteilung des Raumes durch Bassins von ELI nach dem QTAIM-Prinzip ist vollständig, jedoch besitzen die Regionen verschiedenen Charakter. Das Innere der Atome wird in Regionen zerlegt, denen atomare Schalen zugeordnet werden können (atomare Schalenstruktur im Ortsraum). Der Außenbereich, auch Valenzbereich genannt, wird in Regionen für Bindungen und freie Elektronenpaare (lone pairs) aufgeteilt, was der chemischen Beschreibung der Bindungssituation dient. In vielen klassischen Fällen wird hier direkt das Lewis’sche Modell bestätigt, was zum Erfolg dieses Verfahrens maßgeblich beigetragen hat. Überlagert man nun beide Raum-Aufteilungen, so erhält man Informationen, wie die Bindungsregionen zwischen den QTAIM-Atomen aufgeteilt werden, was als Maß für die Bindungspolarität herangezogen werden kann.
Verallgemeinerung der 8–N-Regel für heteropolare Bindungen
. Die grünen und roten Verbindungen zeigen die beiden sich durchdringenden X–Z- und Y–Z-Zinkblende-Partialstrukturen auf. Die 8-fache Koordination des blau gezeichneten Atoms ZHC im X4Y4-Würfel ist hervorgehoben.")

Die genannten Verfahren wurden von Wissenschaftlern der Gruppe „Chemische Bindung“ verwendet (Forschungsbereich Chemische Metallkunde), um eine Lösung für ein altes Problem der auf der Lewis’schen Okettregel basierenden 8−N-Bindungsregel mit polaren Bindungen in halbleitenden intermetallischen Phasen vorzuschlagen [3]. Als Studienobjekt wurden zunächst exemplarisch die Nowotny-Juza-Phasen mit 8 Valenzelektronen im MgAgAs-Strukturtyp herangezogen. Diese Verbindungen besitzen 8 Valenzelektronen und sind elektrisch halbleitend. Die Bindungssituation des elektronegativsten Atoms Z, welches 8-fach würfelförmig von seinen nächsten Nachbaratomen umgeben ist (HC-Position), spielt hier eine entscheidende Rolle. Diese X4Y4 hetero-Würfel (HC) werden aus zwei ineinander gestellten ZX4 und ZY4-Tetraedern gebildet, die jeweils eine Zinkblende-Partialstruktur aufbauen (Abb. 1).
Die Untersuchungen zeigen, dass in den Verbindungen LiMgE15 (E15: ein Element der Stickstoffgruppe auf der HC-Position) die Bindungen E15–Mg4 und E15–Li4 beide stark ionisch sind, in Verbindungen LiE13E14 aber polar-kovalent in einer Zinkblende-Partialstruktur E13–E14 und ionisch in der anderen E14–Li4. Wären beide Partialstrukturen vollständig kovalent, müsste ein metallischer Leiter resultieren, was in keinem der untersuchten Fälle eintritt. Die ionische Li–Si Bindung und die polar-kovalente Al–Si-Partialstruktur in LiAlSi wird typischerweise mit der Formel Li+1[AlSi]–1 angedeutet, was durch die Lewis-Formel impliziert, dass Al eine negative Formalladung zugewiesen wird. Tatsächlich ist die berechnete Ladungsverteilung im Polyanion aber eher mit effektiven Ladungen gemäß [Al1+Si2–]1– charakterisiert. Es stellt sich hier die Frage, ob die quantenchemisch berechneten effektiven Ladungen mit den formalen Ladungen des Pseudoatomkonzeptes im Rahmen der 8–N-Regel vereinbar sind, schließlich sollte eine Si Spezies der Formalladung Si–2 hiernach 2 vollständig kovalente Bindungen ausbilden und zwei freie Elektronenpaare (lone pairs) aufweisen.
 und abgeleiteter Anzahl kovalenter Bindungen NkovB(E), gemäß NkovB = 8 – Nval(E). Rechts: neuartige Bindungsbeziehung (symbolisiert durch Rautenpfeil) zwischen klassischer Zintl-Phase NaP mit 2 homopolaren P–P-Bindungen pro P-Atom und LiAlSi mit 4 heteropolaren Al–Si-Bindungen pro Si-Atom.")

Die vorgeschlagene Lösung besagt, dass die Anzahl der kovalenten Bindungen von der effektiven Ladung des Teilchens mit der höchsten Elektronegativität vorgegeben wird, Si2– also insgesamt 2 kovalente Bindungen mit 4 Al-Atomen ausbildet. Da jede 2-Elektronen-Bindung noch jeweils ein Elektron enthält, welches nicht mit dem Al-Nachbarn geteilt wird, also einen versteckten lone pair-Anteil am Si-Atom besitzt, ist der Kovalenzgrad jeder der 4 Bindungen nur 0,5, und Si erhält die effektive Ladung 2–, Al die Ladung 1+. Die effektiven Ladungen der Atome werden hierbei mittels der QTAIM-Methode bestimmt, die Aufteilung der Bindungselektronen auf die beiden Bindungspartner durch die Überlagerung der Elektronendichte- (QTAIM) und der ELI-Raumaufteilung, also alle Größen aus der quantenchemischen Berechnung. Die erarbeitete Modellvorstellung wurde verallgemeinert für alle Hauptgruppenverbindungen, die der 8–N-Regel gehorchen, und exemplarisch angewendet nicht nur auf eine Reihe von Verbindungen des MgAgAs- und des Zinkblende-Typs, sondern auch auf klassische Zintlphasen (Abb. 2, links). Es ergeben sich hierdurch auch neuartige Bindungsbeziehungen zwischen Atomen mit homopolaren Wechselwirkungen und freien lone pairs und solchen mit heteropolaren Wechselwirkungen und versteckten lone pairs (Abb. 2, rechts) [3].
Atomlagen-Präferenz bei Hauptgruppen- und Übergangsmetallverbindungen des MgAgAs-Typs
Verbindungen des MgAgAs-Typs existieren auch mit Übergangsmetallen in der Zusammensetzung T’TE, wobei T‘ ein frühes Übergangsmetallelement der Gruppen 3–5 darstellt, T ein spätes Übergangsmetallelement der Gruppen 8–10 und das Hauptgruppenelement E vornehmlich aus den Gruppen 14 und 15 stammt. Im häufigsten Fall ist eine Anzahl von 18 Valenzelektronen zu finden, was typischerweise zu unmagnetischem, halbleitendem Verhalten führt. Auffälliger Weise besetzt bei den T’TE-Verbindungen das späte Übergangsmetall die heterokubische Lage, während bei den A’AE-Verbindungen das späte Hauptgruppenelement E diese Position einnimmt. Die Ursache für diese unterschiedliche Lagen-Präferenz wurde mittels quantenchemischer Positionsraum-Werkzeuge untersucht, die eine direkte Verknüpfung zur Energetik der Strukturen besitzen. Im Rahmen der IQA-Methode (Interagierende Quanten-Atome vom QTAIM-Typ) kann die Gesamtenergie eines chemischen Systems in eine Summe von inneratomaren Deformationsenergien und zwischenatomaren Wechselwirkungsenergien umgeschrieben werden. Die Wechselwirkungsenergie setzt sich aus einem klassisch anmutenden elektrostatischen Term und einem kovalenten Austausch-Term zusammen. Der Ansatz ist für Moleküle bereits verfügbar und wird dort mit großem Erfolg angewendet [4]. Für Festkörper wird derzeit noch an der technischen Umsetzung gearbeitet, sodass diese Methodik in voller Präzision dort noch nicht zur Verfügung steht. Stattdessen wurden Punktladungsnäherungen für die beteiligten exakten Größen durchgeführt. Der klassische elektrostatische Term, der im vorliegenden Fall die ionischen Wechselwirkungen quantifiziert, wurde hierbei durch die QTAIM-Madelung-Energie angenähert, das heißt einer klassischen Formel für die Madelung-Energie, die jedoch die QTAIM-Atom-Ladungen verwendet. Zur Spezifizierung der kovalenten Wechselwirkung zwischen den Atomen wurden die zwischenatomaren Delokalisierungsindizes berechnet, die ein Maß für die effektive Bindungsordnung darstellen. Sie skalieren in der Punktladungsnäherung linear mit der interatomaren Austauschenergie. Die Berechnung von Delokalisierungsindizes für Festkörper wurde erstmals durch Wissenschaftler der Gruppe „Chemische Bindung“ vorgestellt [5]. Mit diesem Instrumentarium konnte klar gezeigt werden, dass die Lagepräferenz bei den Hauptgruppenverbindungen von der Madelung-Energie dominiert wird, wobei hier die Verteilung der effektiven Ladungen zu besonders günstigen ionischen Wechselwirkungen führt. Im Unterschied dazu liegt bei den Übergangsmetallverbindungen in den meisten Fällen eine ungünstigere Ladungsverteilung vor, die insgesamt großen kovalenten Beiträge werden hier besonders wichtig und strukturbestimmend. Von besonderer Bedeutung ist hierbei, dass die kovalenten Wechselwirkungen zwischen nächsten Nachbarn nicht nur in der T–E Zinkblende-Partialstruktur vorzufinden sind, sondern auch in vergleichbarem Ausmaß in der T–T’-Partialstruktur [6].
Von Konzepten zu Vorhersagen
 für die Atome T9 = Ir und T10 = Pt der 6. Periode am Beispiel der Verbindungen VT9Ge und HfT10Ge mit den jeweiligen Gitterparametern aGitter. Die kovalenten Bindungsenergiebeiträge –Vx sind für nächste Nachbarn (−Vx(nn)) und zweitnächste Nachbarn (−Vx(2nn)) aufgeschlüsselt, die ionischen Anteile werden durch die QTAIM-Madelung-Energie −EMQTAIM wiedergegeben.")
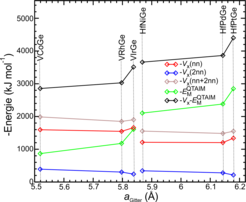
Im nächsten Schritt wurde das beschriebene Instrumentarium dazu verwendet, Bindungs-Trends in 18-Valenzelektronen T’TE-Verbindungen mit MgAgAs-Struktur zu untersuchen und Möglichkeiten für Strukturvorhersagen zu erarbeiten. Es zeigte sich, dass es zu einer gegenseitigen Verstärkung von Kovalenz und Ionizität führt, wenn das späte Übergangsmetall aus den höheren Perioden stammt, speziell, wenn es der 6. Periode angehört. Ursachen sind hier die bessere Ladungsverteilung im Sinne der Madelung-Energie, die Erhöhung der Elektronenzahl in der ersten Koordinationssphäre sowie eine Verschiebung von kovalenten Wechselwirkungen von der zweiten in die erste Koordinationssphäre. Daraufhin wurde die strukturelle Phasengrenze zwischen dem MgAgAs- und dem TiNiSi-Strukturtyp analysiert. Sie liegt bei den T4T10Ge- und T5T9Ge-Verbindungen, wo 14 der 16 bekannten Vertreter den TiNiSi-Typ ausbilden. In diesem Szenario sollten die sechs ausgesuchten Verbindungen T4PtGe (T4= Ti, Zr, Hf) und T5IrGe (T5 = V, Nb, Ta) aufgrund ihrer optimierten Bindungseigenschaften im MgAgAs-Typ besonders wettbewerbsfähig gegen den TiNiSi-Typ sein (Abb. 3).
Die experimentelle Überprüfung dieser Vorhersage ergab, dass 4 von den 6 Kandidaten tatsächlich den MgAgAs-Typ aufwiesen. Zwei dieser Phasen waren bereits bekannt, TiPtGe aus eigenen Arbeiten [7] und TaIrGe, welches zur gleichen Zeit mittels „ab initio Thermodynamik“ vorhergesagt und anschließend synthetisiert worden war [8]. Somit wurden zwei Drittel der vorhergesagten Verbindungen experimentell bestätigt. Bemerkenswert ist, dass die beiden neu entdeckten Phasen, VIrGe und LT-HfPtGe, nur in einem begrenzten Temperaturfenster existieren, und ohne die gezielte Synthese wohl weiter übersehen worden wären [9].


Beispielsweise ist beim Aufheizen der aus der Schmelze hergestellten Probe von HfPtGe im Kalorimeter das sehr kleine Signal bei 1010°C nur bei genauem Hinsehen zu erkennen (Abb. 4, Mitte unten). Dieser Effekt signalisiert den Phasenübergang zwischen der bekannten Hochtemperatur- und der Tieftemperatur-Modifikation mit vorhergesagtem MgAsAs-Strukturtyp. Um den mengenmäßigen Anteil der Kristalle mit MgAgAs-Struktur zu erhöhen, wurde die Probe für 30 Tage konstant bei 900°C erwärmt. Danach zeigt das aufgenommene Röntgendiffraktogramm (Abb. 4, links) die vollständige Umwandlung in die Tief-Temperaturmodifikation vom MgAgAs-Typ an. Die von dieser Probe dann nachfolgend aufgenommene Aufheizkurve im Kalorimeter weist nun ein deutlicheres Signal für die Umwandlung in die Hochtemperatur-Modifikation vom TiNiSi-Typ auf (Abb. 4, Mitte unten). Es zeigt sich also wieder, dass Manches unter der Oberfläche schlummert, das erst gefunden wird, wenn gezielt danach gesucht wird. Die chemische Bindungsanalyse im Ortsraum hat im vorliegenden Fall diese gezielte Suche initiiert.
Literaturhinweise
100 Years Old and Getting Stronger